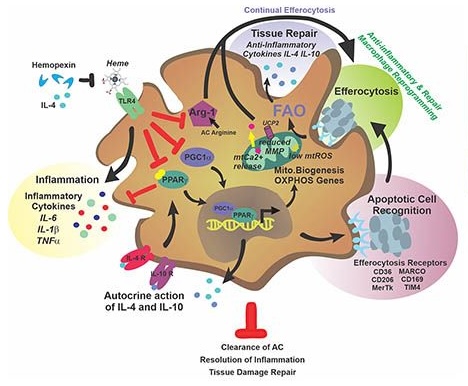

Sickle cell disease (SCD) is hallmarked by an underlying chronic inflammatory condition, which is contributed by heme-activated pro-inflammatory macrophages. While previous studies addressed heme ability to stimulate macrophage inflammatory skewing through TLR4/ROS signaling, how heme alters cell functional properties remains unexplored. Macrophage-mediated immune cell recruitment and apoptotic cell (AC) clearance are relevant in the context of SCD, where tissue damage, cell apoptosis and inflammation occur due to vasoocclusive episodes, hypoxia and ischemic injury. Here we show that heme strongly alters macrophage functional response to AC damage by exacerbating immune cell recruitment and impairing cell efferocytic capacity. In SCD, heme-driven excessive leukocyte influx and defective efferocytosis contribute to exacerbated tissue damage and sustained inflammation. Mechanistically, these events depend on heme-mediated activation of TLR4 signaling and suppression of the transcription factor PPARg and its coactivator PGC1a. These changes reduce efferocytic receptor expression and promote mitochondrial remodeling, resulting in a coordinated functional and metabolic reprogramming of macrophages. Overall, this results in limited AC engulfment, impaired metabolic shift to mitochondrial fatty acid b-oxidation and ultimately reduced secretion of the anti-inflammatory cytokines IL-4 and IL-10, with consequent inhibition of continual efferocytosis, resolution of inflammation and tissue repair. We further demonstrate that impaired phagocytic capacity is recapitulated by macrophage exposure to sickle patients’plasma and improved by hemopexin-mediated heme scavenging, PPARg agonists or IL-4 exposure through functional and metabolic macrophage rewiring. Our data indicate that therapeutic improvement of heme-altered macrophage functional properties via heme scavenging or PGC1a/PPARg modulation significantly ameliorate tissue damage associated with SCD pathophysiology.
This work was supported by University of Utah Center of Excellence in Hematology (CIHD) U54 DK110858 Pilot and Feasibility Grant to F.V. and the Metabolomics Core at the University of Utah School of Medicine.